LFD, PCR, PPV, TLA
People are very interested in the positive predictive value of lateral flow tests. That is, if you receive a positive result from the test, how likely is it that you truly are infected? Personally I think that in terms of measuring whether these tests are helpful to society, this metric is actually not terribly useful - since it is strongly affected by how much virus is circulating in the population. That means that the metric implies that lateral flow testing is useless in countries with very low numbers of cases – but actually LFDs have exactly the same effect on R whatever the level of virus in the population, and the same number of absolute false positives.
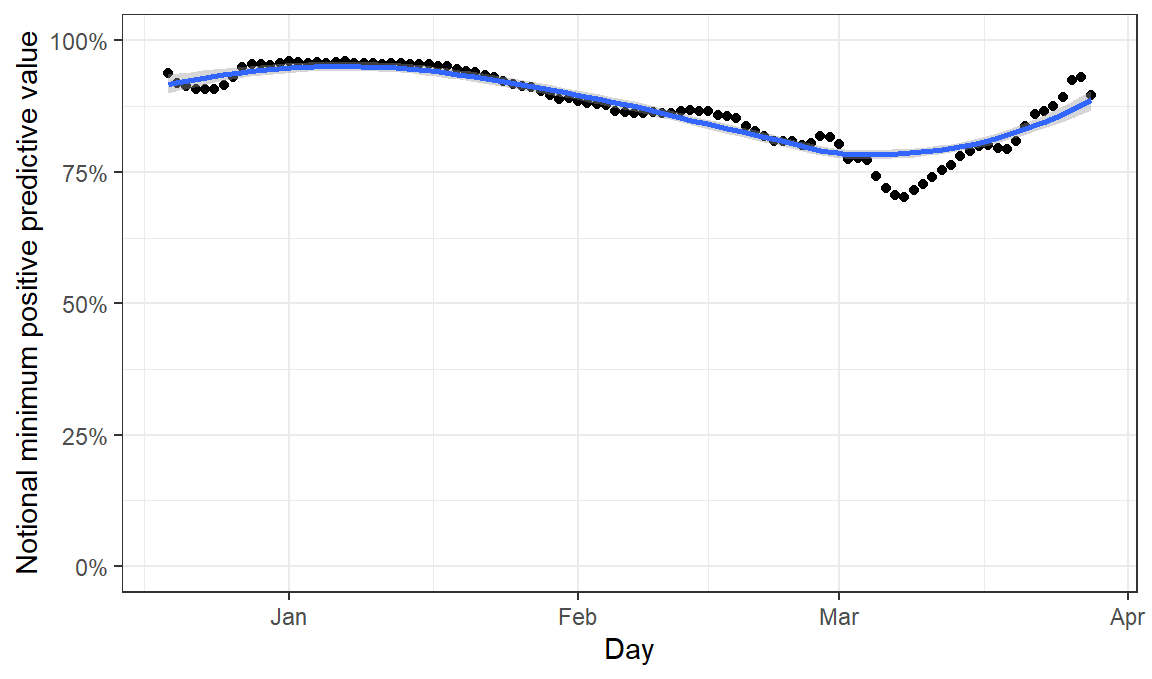
Nevertheless, it is understandable that people want a test to ideally give them the right answer about their own status. They are now helped by the fact that the government says that all LFD positives should be followed up by confirmatory PCR testing, which both helps an individual to confirm their status, and also has the potential to provide us all with information about the general reliability of these tests. Information has not been released systematically on what proportion of PCR retests confirm LFD results, but today the coronavirus dashboard has changed to exclude cases which were retested and gave negative results. Therefore by comparing today’s results with yesterdays we can get a sense of this metric and calculate the minimum possible PPV, assuming that the PCR retest has 100% sensitivity. These sorts of analyses have already been described by Oliver Johnson and Alex Selby amongst others.
Here is my quick look in R
before = read_csv("https://api.coronavirus.data.gov.uk/v2/data?areaType=nation&areaCode=E92000001&metric=newCasesLFDOnlyBySpecimenDate&metric=changeInNewCasesBySpecimenDate&format=csv&release=2021-04-08&metric=newCasesLFDConfirmedPCRBySpecimenDate")
#> Parsed with column specification:
#> cols(
#> date = col_date(format = ""),
#> areaType = col_character(),
#> areaCode = col_character(),
#> areaName = col_character(),
#> newCasesLFDOnlyBySpecimenDate = col_double(),
#> changeInNewCasesBySpecimenDate = col_double(),
#> newCasesLFDConfirmedPCRBySpecimenDate = col_double()
#> )
after = read_csv("https://api.coronavirus.data.gov.uk/v2/data?areaType=nation&areaCode=E92000001&metric=newCasesLFDOnlyBySpecimenDate&metric=changeInNewCasesBySpecimenDate&format=csv&release=2021-04-09&metric=newCasesLFDConfirmedPCRBySpecimenDate")
#> Parsed with column specification:
#> cols(
#> date = col_date(format = ""),
#> areaType = col_character(),
#> areaCode = col_character(),
#> areaName = col_character(),
#> newCasesLFDOnlyBySpecimenDate = col_double(),
#> changeInNewCasesBySpecimenDate = col_double(),
#> newCasesLFDConfirmedPCRBySpecimenDate = col_double()
#> )
library(zoo)
#>
#> Attaching package: 'zoo'
#> The following objects are masked from 'package:base':
#>
#> as.Date, as.Date.numeric
both = inner_join(before,after,by="date", suffix=c("_before","_after")) %>% filter(date>"2020-12-15")%>% filter(date<"2021-04-01")
both= both %>% mutate(notional_false_positives = newCasesLFDOnlyBySpecimenDate_before - newCasesLFDOnlyBySpecimenDate_after - (newCasesLFDConfirmedPCRBySpecimenDate_before - newCasesLFDConfirmedPCRBySpecimenDate_after) ) %>% arrange(date) %>% mutate(notional_false_positives=rollsum(notional_false_positives,7,na.pad=T),newCasesLFDConfirmedPCRBySpecimenDate_after=rollsum(newCasesLFDConfirmedPCRBySpecimenDate_after,7,na.pad=T)) %>% mutate( notional_proportion_of_positives_false = notional_false_positives / (notional_false_positives+ newCasesLFDConfirmedPCRBySpecimenDate_after ) )%>% filter(notional_false_positives>0)
ggplot(both,aes(x=date,y=1-notional_proportion_of_positives_false))+geom_point()+geom_smooth()+coord_cartesian(ylim=c(0,1))+labs(x="Day",y="Notional minimum positive predictive value")+scale_y_continuous(label=scales::percent)+theme_bw()
#> `geom_smooth()` using method = 'loess' and formula 'y ~ x'
Theo Sanderson
Assistant Professor
Biologist developing tools to scale pathogen genetics.