Biography
I am an Assistant Professor at the London School of Hygiene and Tropical Medicine where I lead a research group working on large-scale viral genomics.
- Viral Genomics
- Phylogenetics
- Malaria
- Open Source Hardware
- Machine Learning
-
PhD in Malaria Genetics, 2016
Wellcome Sanger Institute
-
BA Natural Sciences, 2011
University of Cambridge
Selected Publications
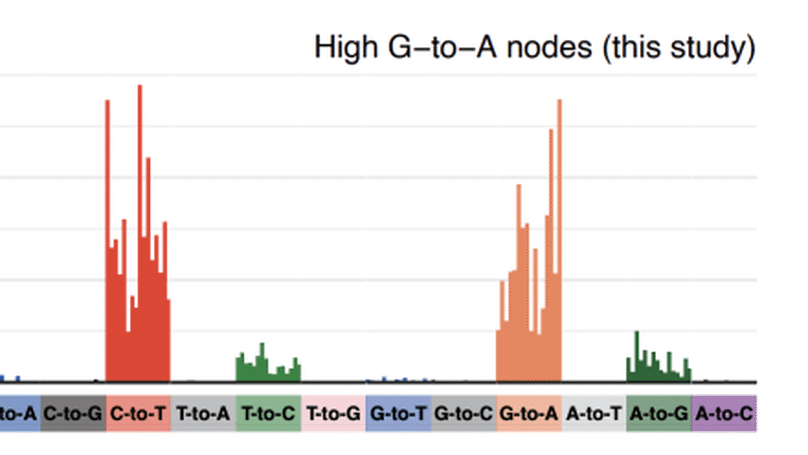
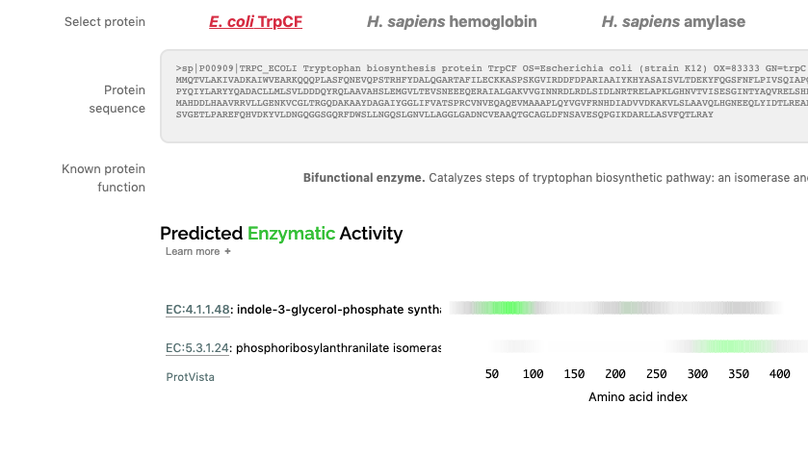
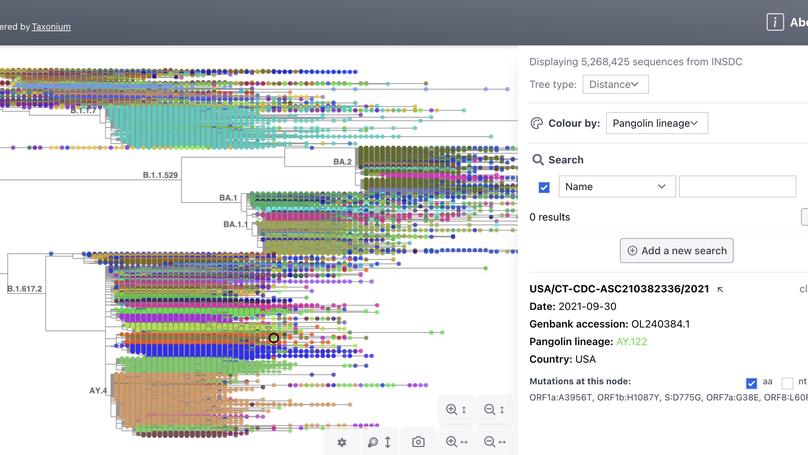
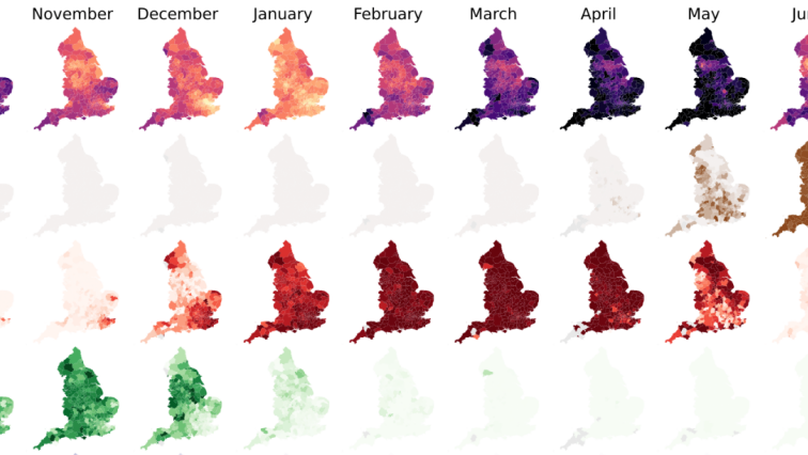
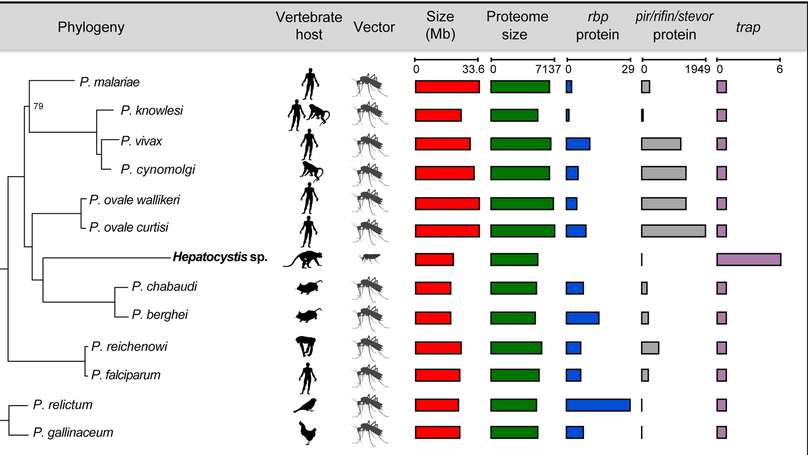
This work began when I did a BLAST search for a malaria parasite gene, and saw a closely matching gene that claimed to be from a monkey. When I investigated further I found that this “monkey genome” contained substantial contamination from a genus of parasite called Hepatocystis that had been lurking in the monkey’s blood. The identification of the first substantial genomic data from this genus, which I initially described in a blog post, triggered a collaborative project between the originators of the data, former colleagues at the Sanger Institute, and myself to characterise this genome revealing the genomic basis of this parasite’s unique biology.
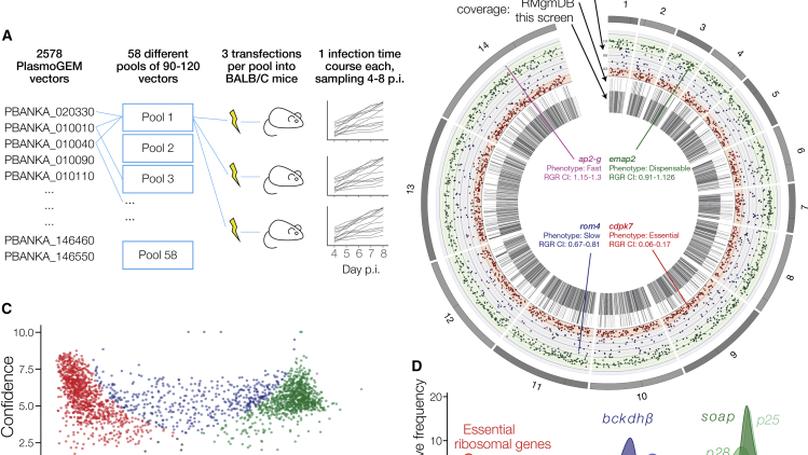
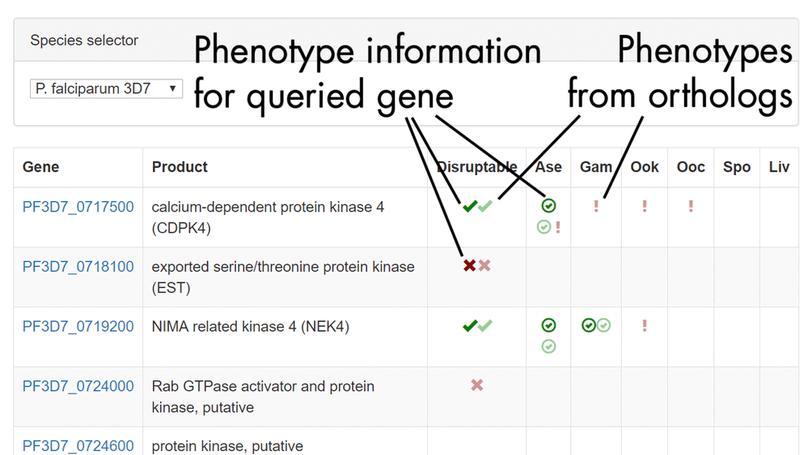
In this work we conducted the first genome-scale genetic screen in a malaria parasite. We found that malaria parasites have require a higher proportion of their genome for normal growth compared to any other eukaryote previously screened. I led the analysis portion of this work, including building the dashboard used by the community to access our phenotype data.
Recent Publications
Recent Posts
Positions held
Contact
- theo@theo.io
- London School of Hygiene and Tropical Medicine, London, WC1E 7HT